Decision Making and Loops -- Additional Detail
Overview
Class Date: 9/16/2025 -- On Your Own
Teaching: 90 min
Exercises: 30 minQuestions
What operators and functions are available for running test in
ifstatements?Are there different indexing strategies for different situations using a
forloop?Should I use a loop or an
applystatement?Objectives
Know the bredth of options for running test (aka asking questions) in
ifstatements.Practice applying
ifstatements in different situations.Understand how R interprets simple operations (e.g. addition) when applied to data vectors.
Use either the elements or the position in a vector as the primary index in a
forloop.Trace changes to a loop variable as the loop runs.
Trace changes to other variables as they are updated by a
forloop.Understand the advanatages and disadvantages to using apply functions vs. for loops.
Practice applying
forloops in different situations.
On Your Own
Conditional statements – additional detail
We covered the basics of using if and else statement In Class. These make up a central tool in coding. There are a lot of ways to write the logical tests in if statements to ask specific and complex questions about your data and metadata. Here is a useful reference to help keep the options straight:
Common operators and functions used in
ifstatmentsConditional tests can be as complicated as you need them to be. Here is set of operators and functions commonly used in
ifstatement tests:Relational operators:
<= less than>= greater than<== less than or equal to>== greater than or equal to=== exactly equal to!== not equal to%in%= is present in (used to ask if the value(s) on the left is present in the vector/matrix on the right)Logical operators:
!= NOT (changesTRUEtoFALSEand vice versa)&= element-wise AND (both are true; outputs vector for vector input comparing elements)&&= logical AND (both are true; only considers first element of a vector)|= element-wise OR (one or both are true; outputs vector for vector input comparing elements)||= logical OR (both are true; only considers first index of a vector)xor(x,y)= element-wise exclusive OR (either are true, but not both; outputs vector for vector input comparing elements)Functions:
all()= are all entriesTRUE?any()= is at least one elementTRUE?is.na()= is the input valueNA?is.<data type>()= does the input object belong to class<data type>?is.finite()= is the input value finite?is.infinite()= is the input value infinite?
Beyond that, the best way to learn how to use if statements effectively is to practice applying them to different situations. Below are a series of exercises to get you started.
Exercises – conditional statements
Histograms Instead
One of your collaborators prefers to see the distributions of larger vectors as a histogram instead of as a boxplot. In order to choose between a histogram and a boxplot we will edit the function
plot_dist()> that we wrote In Class and add an additional argumentuse_boxplot.
By default we will setuse_boxplottoTRUEwhich will create a boxplot when the vector is longer thanthreshold.When
use_boxplotis set toFALSE,plot_dist()will instead plot a histogram for larger vectors. As before, if the length of the vector is shorter thanthreshold,plot_dist()will create a stripchart (allowing users to define what is meant by “larger”).A histogram is made with the
hist()command in R. Here is how we wantplot_dist()to behave:dat <- read.csv("data/inflammation-01.csv", header = FALSE) plot_dist(dat[, 10], threshold = 10, use_boxplot = TRUE) # day (column) 10 - create boxplot
plot_dist(dat[, 10], threshold = 10, use_boxplot = FALSE) # day (column) 10 - create histogram
plot_dist(dat[1:5, 10], threshold = 10) # samples (rows) 1-5 on day (column) 10
Solution
# Copy and paste the plot_dist() function from In Class # and add "use_boxplot" as an argument with a default value of TRUE plot_dist <- function(x, threshold, use_boxplot = TRUE) { # if the vector is above threshold, use boxplot if requested if (length(x) > threshold && use_boxplot) { boxplot(x) # if a boxplot is not requested, use histogram instead } else if (length(x) > threshold && !use_boxplot) { hist(x) # if the vector is below the length threshold, use a stripchart } else { stripchart(x, vert=T) } }
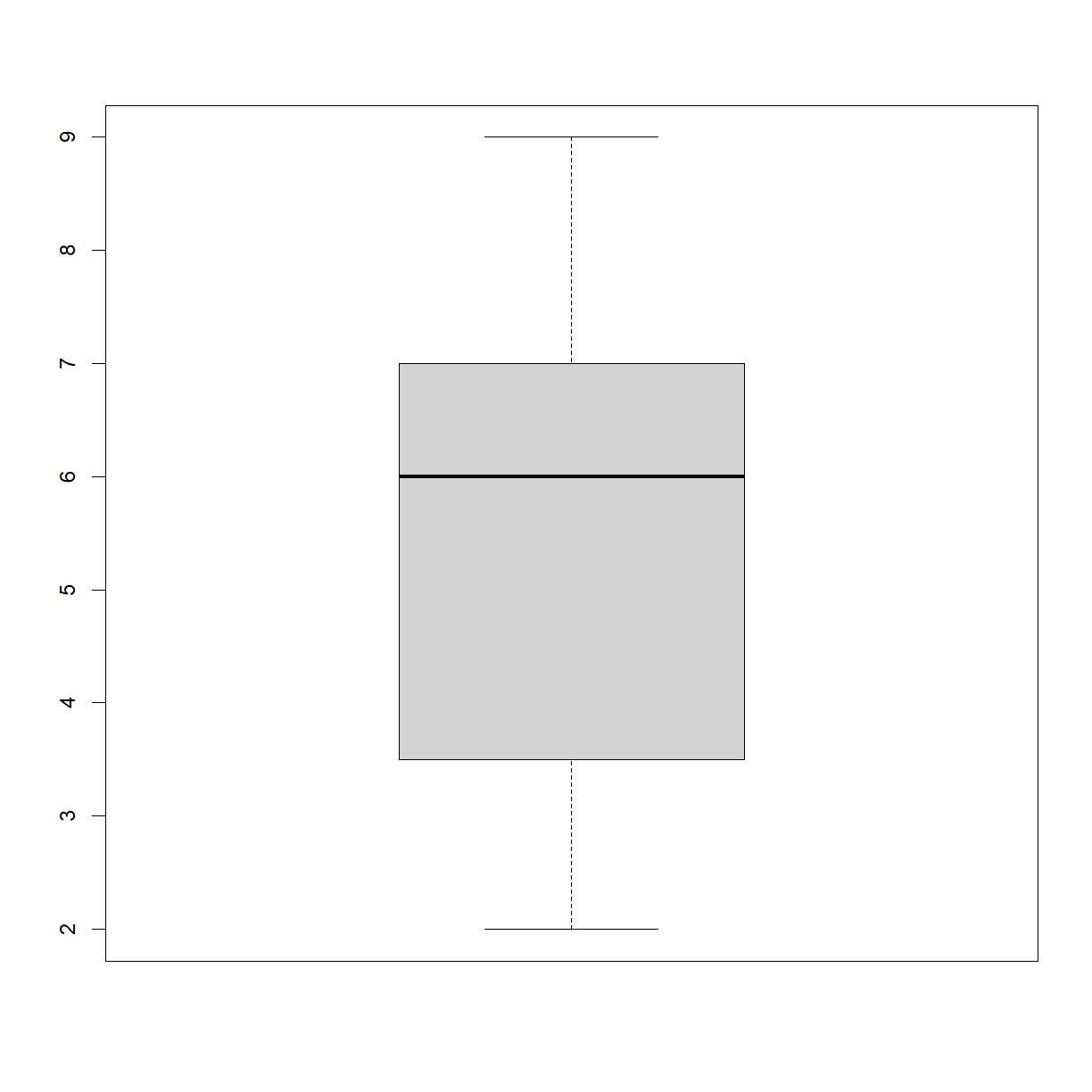
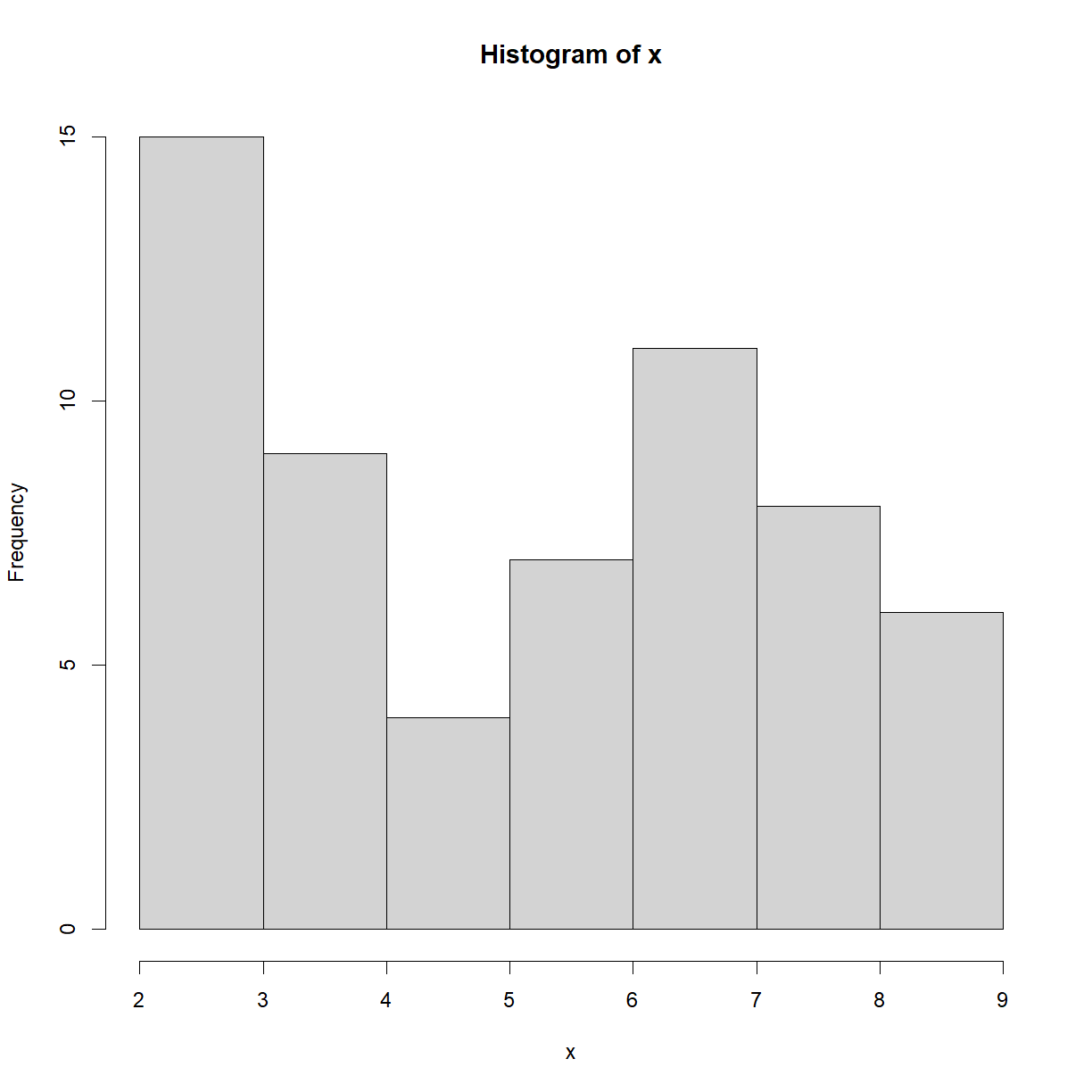
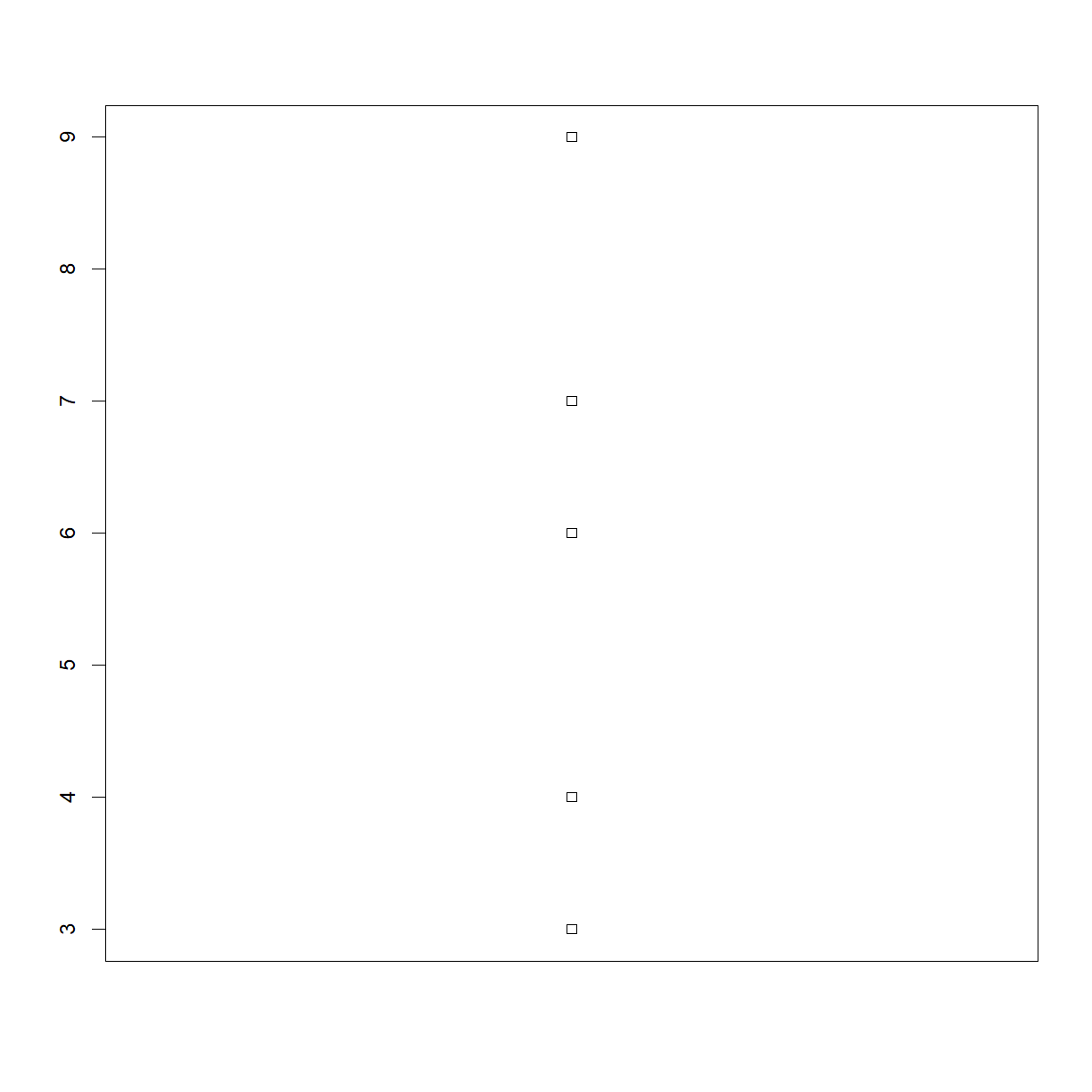
Combining concepts – segretating flowers by petal size
In the
irisdataset, we want to categorize each flower based on size. Copy theirisdataset:iris2 <- iris
and add a column that indicates whether that specific flower is a member of it’s species with “large” petals (
Petal.Length> species average ofPetal.Length) or “small” petals (Petal.Length<= species average ofPetal.Length).To accomplish this goal, you will need to write a script with several steps combining concepts from different lessons. Plan out the necessary steps and how to accomplish each step with concepts covered so far. Try to find answers to questions using forums or other online resources; however, there are a few progressive hints below if you get really stuck.
Hint 1 – Planning your script
One approach to this problem is to complete three steps:
- Calculate the average
Petal.Lengthfor each species and store the values in a searchable format (e.g. data frame withSpeciesname in one column, average `Petal.Length in the other).- Identify the correct average
Petal.Lengthvalue for each flower based based on it’s species.- Compare the
Petal.Lengthfor each flower to the corresponding average speciesPetal.Lengthand output eitehr “large” or “small”.Hint 2
The
aggregate()function is useful for completing basic function calls (e.g.mean())across different subsets of a dataset.Hint 3
The
match()function allows you to take a vector of values, and look up the position in a second vector that matches each element of the first. The output can be used to index the second list (or another of the same length) to return corresponding values.Hint 4
The
ifelse()function is a vectorized version of anifandelsestatement that allows you to return one of two values based on a relational comparison that returns a logical statement.Solution
The first step is to calculate the average
Petal.Lengthfor each species. An efficient way to do this is to use theaggregate()function:petal.length.means <- aggregate(Petal.Length~Species, data=iris2, FUN=mean) petal.length.meansSpecies Petal.Length 1 setosa 1.462 2 versicolor 4.260 3 virginica 5.552
The second step is to determine which of these mean values is the correct one to use for comparison for each flower (i.e. which species does the flower belong to). To do this, we can create a vector that contains the correct mean comparison value for each flower in the
irisdata frame. Thematch()function allows us to look up the mean value in thepetal.length.meansdata frame based on theSpeciesfor each flower in theirisdata frame:# first look up the index species.index <- match(iris$Species,petal.length.means$Species) species.index[1] 1 1 1 1 1 1 1 1 1 1 1 1 1 1 1 1 1 1 1 1 1 1 1 1 1 1 1 1 1 1 1 1 1 1 1 1 1 [38] 1 1 1 1 1 1 1 1 1 1 1 1 1 2 2 2 2 2 2 2 2 2 2 2 2 2 2 2 2 2 2 2 2 2 2 2 2 [75] 2 2 2 2 2 2 2 2 2 2 2 2 2 2 2 2 2 2 2 2 2 2 2 2 2 2 3 3 3 3 3 3 3 3 3 3 3 [112] 3 3 3 3 3 3 3 3 3 3 3 3 3 3 3 3 3 3 3 3 3 3 3 3 3 3 3 3 3 3 3 3 3 3 3 3 3 [149] 3 3# and use it to pull the mean value mean.comparison <- petal.length.means$Petal.Length[species.index] mean.comparison[1] 1.462 1.462 1.462 1.462 1.462 1.462 1.462 1.462 1.462 1.462 1.462 1.462 [13] 1.462 1.462 1.462 1.462 1.462 1.462 1.462 1.462 1.462 1.462 1.462 1.462 [25] 1.462 1.462 1.462 1.462 1.462 1.462 1.462 1.462 1.462 1.462 1.462 1.462 [37] 1.462 1.462 1.462 1.462 1.462 1.462 1.462 1.462 1.462 1.462 1.462 1.462 [49] 1.462 1.462 4.260 4.260 4.260 4.260 4.260 4.260 4.260 4.260 4.260 4.260 [61] 4.260 4.260 4.260 4.260 4.260 4.260 4.260 4.260 4.260 4.260 4.260 4.260 [73] 4.260 4.260 4.260 4.260 4.260 4.260 4.260 4.260 4.260 4.260 4.260 4.260 [85] 4.260 4.260 4.260 4.260 4.260 4.260 4.260 4.260 4.260 4.260 4.260 4.260 [97] 4.260 4.260 4.260 4.260 5.552 5.552 5.552 5.552 5.552 5.552 5.552 5.552 [109] 5.552 5.552 5.552 5.552 5.552 5.552 5.552 5.552 5.552 5.552 5.552 5.552 [121] 5.552 5.552 5.552 5.552 5.552 5.552 5.552 5.552 5.552 5.552 5.552 5.552 [133] 5.552 5.552 5.552 5.552 5.552 5.552 5.552 5.552 5.552 5.552 5.552 5.552 [145] 5.552 5.552 5.552 5.552 5.552 5.552# this can all be done in one step: mean.comparison <- petal.length.means$Petal.Length[match(iris$Species,petal.length.means$Species)]
Finally, we use the vector of mean values to compare each flower’s
Petal.Lengthto the species averagePetal.Lengthusingifelse()and assign “large” if the flower is larger or “small” if not.iris2$Species.Petal.Group <- ifelse(iris2$Petal.Length > mean.comparison, "large", "small") head(iris2)Sepal.Length Sepal.Width Petal.Length Petal.Width Species Species.Petal.Group 1 5.1 3.5 1.4 0.2 setosa small 2 4.9 3.0 1.4 0.2 setosa small 3 4.7 3.2 1.3 0.2 setosa small 4 4.6 3.1 1.5 0.2 setosa large 5 5.0 3.6 1.4 0.2 setosa small 6 5.4 3.9 1.7 0.4 setosa large
Using for loops – additional detail
Setting up the loop variable in for loops – two common approaches
There is a subtle decision to make when designing a for loop. In Class we demonstrated how to use the loop variable to step through a collection of objects and repeat some desired task. R updates the loop variable during each loop (aka each iteration) by reassigning the value of the loop variable to the next object sequentially in the collection. However, we can use different properties of the collection to define the value assigned to the loop variable during each iteration. The two most common are to have the loop variable take on, in sequence:
- the value of the object contained within each element, or
- the position (i.e. index) of the element within the collection
Each is perfectly acceptable, and useful in different situations.
Iterating by element
Updating the loop variable to take on the value of the next element in the collection during each iteration is the version we used in in class. To do this, we define the loop variable using the format for(item in collection). To illustrate what the loop is doing with the loop variable during each iteration, let’s write a self-referential for loop that just prints the value of the loop variable once each loop. We will use the first 6 letters in the alphabet as our collection:
# define our collection
collection <- letters[1:6]
collection
[1] "a" "b" "c" "d" "e" "f"
# iterate through the collection and print each element
for(item in collection) {
print(item)
}
[1] "a"
[1] "b"
[1] "c"
[1] "d"
[1] "e"
[1] "f"
We can now use the values contained within our collection in operations within the loop. As an arbitrary example, we can add the letters to a growing string and print that string:
string <- "" # initialize empty string
for(item in collection) {
print(item) # print current item
string <- paste0(string,item) # add letter to current string
print(string) # print current string
}
[1] "a"
[1] "a"
[1] "b"
[1] "ab"
[1] "c"
[1] "abc"
[1] "d"
[1] "abcd"
[1] "e"
[1] "abcde"
[1] "f"
[1] "abcdef"
This structure for the for loop is important when all you need to manipulate within the loop are the values contained within the collection.
Iterating by position
Often times, there will be a good reason to know the current position (or index) in the collection during a given loop, rather than the contents of the collection at that position. The index allows us to do a variety of things, including index other objects with the same dimensions or provide the user with a progress update for more time consuming jobs.
For positional indexing, we can use the format for(1:length(collection)). Alternatively, we can use the seq_along() function, which is a version of seq that uses arguments from = 1 and by = 1 by default, and the length of the entered object for the to argument. Thus 1:length(collection) produces the same result as seq_along(collection):
collection
[1] "a" "b" "c" "d" "e" "f"
1:length(collection)
[1] 1 2 3 4 5 6
seq_along(collection)
[1] 1 2 3 4 5 6
In essence, we are creating a new collection for the for loop that consists of a list of integers from 1 to the length of the collection. The for loop will step through these integers, in effect using the loop variable too keep count of the number of loops that have progressed. Let’s look at the behavior by replacing the loop variable from our above example with the positional version (and renaming item with i to indicate that we are looking at the index, rather than the contents, of collection:
for(i in 1:length(collection)) {
print(i)
}
[1] 1
[1] 2
[1] 3
[1] 4
[1] 5
[1] 6
Note that we can still easily grab the contents of collection in each loop by simply indexing it with our loop variable:
for(i in 1:length(collection)) {
print(collection[i])
}
[1] "a"
[1] "b"
[1] "c"
[1] "d"
[1] "e"
[1] "f"
Because we can easily grab the current element of collection in this way, and the current index is more generally useful than the content of the current collection element, I tend to use positional iteration in for loops by default. This one comes down to personal preference.
One place that positional iteration is commonly useful is when you want to use a for loop to repeat some operation on each row of a data frame. Let’s revisit our aneurism data set:
dat <- read.csv("data/sample.csv")
head(dat)
ID Gender Group BloodPressure Age Aneurisms_q1 Aneurisms_q2
1 Sub001 m Control 132 16.0 114 140
2 Sub002 m Treatment2 139 17.2 148 209
3 Sub003 m Treatment2 130 19.5 196 251
4 Sub004 f Treatment1 105 15.7 199 140
5 Sub005 m Treatment1 125 19.9 188 120
6 Sub006 M Treatment2 112 14.3 260 266
Aneurisms_q3 Aneurisms_q4
1 202 237
2 248 248
3 122 177
4 233 220
5 222 228
6 320 294
Recall that in earlier lessons we corrected the capitalization inconsistency in the Gender variable, and also calculated several summary statistics for the quarterly aneurisms counts (total, mean, maximum). We previously did these using a combination of vectorized functions and the apply() function. Let’s look at how we might accomplish the same set of tasks using a for loop:
# first define a new empty column for each new statistic
# that we want to calculate; use NAs to indicate no value
# has yet been calculated
dat$aneurisms_total <- NA
dat$aneurisms_mean <- NA
dat$aneurisms_max <- NA
# set up a for loop to cycle through each row of the data frame
# using the number of rows provided by the dim() function; you
# could alternatively use 1:length(dat$ID) for example
for(i in 1:dim(dat)[1]) {
# first use an if statements to correct lower to upper case Gender
# for the current (ith) entry
if(dat$Gender[i] == "m") {dat$Gender[i] <- "M"} # do nothing if not "m"
if(dat$Gender[i] == "f") {dat$Gender[i] <- "F"} # do nothing if not "f"
# now calculate the current (ith) value for each of the statistics;
# note that we need to include `as.numeric()` to tell R to treat
# the columns of the data frame instead as a numeric vector
dat$aneurisms_total[i] <- sum(as.numeric(dat[i,6:9]))
dat$aneurisms_mean[i] <- sum(as.numeric(dat[i,6:9]))
dat$aneurisms_max[i] <- sum(as.numeric(dat[i,6:9]))
}
# check the result
head(dat)
ID Gender Group BloodPressure Age Aneurisms_q1 Aneurisms_q2
1 Sub001 M Control 132 16.0 114 140
2 Sub002 M Treatment2 139 17.2 148 209
3 Sub003 M Treatment2 130 19.5 196 251
4 Sub004 F Treatment1 105 15.7 199 140
5 Sub005 M Treatment1 125 19.9 188 120
6 Sub006 M Treatment2 112 14.3 260 266
Aneurisms_q3 Aneurisms_q4 aneurisms_total aneurisms_mean aneurisms_max
1 202 237 693 693 693
2 248 248 853 853 853
3 122 177 746 746 746
4 233 220 792 792 792
5 222 228 758 758 758
6 320 294 1140 1140 1140
In this case, the point is more illustrative of how to design a for loop. Using toupper() to correct the Gender column and 3 apply() statements to take care of the statistics would accomplish in 4 lines of code what too 11 in the for loop version. However, there will be times when the analysis you want to conduct is too complex to formulate concisely within an apply() statement or two.
Loops in R are slow (??)
On this topic, you will probably also hear at some point if you keep using R that loops are slow. This is a common refrain in coding forums. In point of fact, loops are not slow if you follow a few guidelines:
- Don’t use a loop when a vectorized alternative exists; unlike
apply, vectorized operations are faster than loops - Don’t grow objects (via
c(),cbind(), etc) during the loop - R has to create a new object and copy across the information just to add a new element or row/column - Instead, pre-allocate an empty object that is large enough to hold the results (e.g. a data frame with the right number of rows and columns, but with
NAor default values pre-loaded) and fill in the individual values during each iteration of theforloop. For instance, in the aneurisms example above, we pre-defined (aka initialized) empty columns in the data frame for the total, mean, and max statistics before starting theforloop.
As an example, we will create a new function called analyze() that will return the mean inflammation per day (column) of each file in the inflammation dataset that we examined earlier. We will start by breaking rule 2, and grow our output by adding new data to a growing data frame using cbind():
analyze <- function(filenames) {
# start by defining the loop parameters
for (f in seq_along(filenames)) {
fdata <- read.csv(filenames[f], header = FALSE)
res <- apply(fdata, 2, mean)
# check if this is the first iteration
if (f == 1) {
# if so, the current result is the only line of output
out <- res
} else {
# If this is not the first iteration, add the result to the
# established result as a new column
out <- cbind(out, res)
}
}
return(out)
}
Note how we add a new column to out during each iteration? This is a cardinal sin of writing a for loop in R.
A much better practice is to create an empty matrix with the right dimensions (rows/columns) to hold the results before starting the for loop. Then we loop over the files as before, but this time we fill in the pre-defined fth column of our results matrix out instead of creating a new column from scratch on the fly. This time there is no copying/growing for R to deal with.
analyze2 <- function(filenames) {
# pre-allocate the right number of rows and columns for the data
out <- matrix(ncol = length(filenames), nrow = 40)
# run the for loop to cycle through the files
for (f in seq_along(filenames)) {
# read the current file
fdata <- read.csv(filenames[f], header = FALSE)
# instead of using cbind, now we just overwrite the row of
# the pre-allocated matrix
out[, f] <- apply(fdata, 2, mean)
}
return(out)
}
Now we can use the function system.time() to find out how long it takes to run each of these functions:
# First, make sure our inflammation file list is defined
inflam.files <- list.files(path = "data",
pattern = "^inflammation-.*.csv$",
full.names = TRUE)
gc() # clean things up first
used (Mb) gc trigger (Mb) max used (Mb)
Ncells 652366 34.9 1336134 71.4 1336134 71.4
Vcells 1217076 9.3 8388608 64.0 2356649 18.0
# one call to each function does not always show the difference. To
# demonstrate the point, we will use a for loop to call function 100
# times in a row to see how long each takes
system.time(avg <- for(i in 1:100) {analyze(inflam.files)})
user system elapsed
1.16 0.22 1.39
system.time(avg <- for(i in 1:100) {analyze2(inflam.files)})
user system elapsed
1.30 0.06 1.36
The system time output indicates the total CPU time used to execute the function with elapsed reflects the total real-world time consumed between when the function was called and when the output was generated. Your results may vary from run to run, but the amount of time used by analyze should always be greater than the time used by analyze2, all else being equal. A few 10ths of a second may not seem much now, but keep in mind that we are running a few simple operations on a few hundred files in total. As the complexity of your analysis and the number of files or data point in your dataset increases, the time difference will increase dramatically. A given analysis taking 6 hours to process vs. 30 hours can be critical to the success of some big-data projects, even before you start to calculate the monetary expense of running large analyses on a super computer. This demonstrates one way to evaluate efficiency when it starts to matter.
Note that apply handles these memory allocation issues for you, but then you have to write the loop part as a function to pass to apply. At its heart, apply is just a for loop with extra convenience.
Organizing Larger Projects
On the topic of large datasets, for larger projects, it is recommended to organize separate parts of the analysis into multiple subdirectories, e.g. one subdirectory for the raw data, one for the code, and one for the results like figures. We have done that here to some extent, putting all of our data files into the subdirectory “data”. For more advice on this topic, you can read A quick guide to organizing computational biology projects by William Stafford Noble.
for or apply?
A for loop is used to apply the same function calls to a collection of objects. As we just demonstrated, the apply family of functions are used in much the same way.
You’ve already used one of the family, apply(). The apply family members include:
apply()- apply over the margins of an array (e.g. the rows or columns of a matrix)lapply()- apply over an object and return listsapply()- apply over an object and return a simplified object (an array) if possiblevapply()- similar tosapply()but you specify the type of object returned by the iterations- Functions provided in external packages, such as
ddply()andadply()in the populardplyrpackage.
Each of these has an argument FUN (or something similar) which takes a function to apply to each element of the object. Instead of looping over filenames and calling analyze, as you did earlier, you could sapply over filenames with FUN = analyze:
sapply(filenames, FUN = analyze)
Deciding whether to use for or one of the apply family is really personal preference. Using an apply family function forces to you encapsulate your operations as a function rather than separate calls with for. for loops are often more intuitive. For several related operations, a for loop will avoid you having to pass in a lot of extra arguments to your function.
You will sometimes see claims that apply functions run faster than a for loop designed to run the same operations. However, this is not generally true, and under the surface the apply functions are just convenient wrappers for a hidden for loops.
Exercises – for loops
Summing Values
Write a function called
total()that calculates the sum of the values in a vector.Yes, R has a built-in function called
sum()that does this for you. Please don’t use it for this exercise. The goal is to understand how this type of function might work under the surface.ex_vec <- c(4, 8, 15, 16, 23, 42) total(ex_vec)[1] 108Solution
total <- function(vec) { # calculates the sum of the values in a vector vec_sum <- 0 for (num in vec) { vec_sum <- vec_sum + num } return(vec_sum) }
Exponentiation
Exponentiation is built into R:
2^4[1] 16Write a function called
expo()that uses a loop to calculate the same result without using the^operator.expo(2, 4)[1] 16Solution
expo <- function(base, power) { result <- 1 for (i in seq(power)) { result <- result * base } return(result) }
Find the Maximum Inflammation Score
Find the file containing the patient with the highest average inflammation score. Print the file name, the patient number (row number) and the value of the maximum average inflammation score.
Tips:
- Use variables to store the maximum average and update it as you go through files and patients.
- You can use nested loops (one loop is inside the other) to go through the files as well as through the patients in each file (every row).
Complete the code below:
filenames <- list.files(path = "data", pattern = "^inflammation-.*.csv$", full.names = TRUE) filename_max <- "" # filename where the maximum average inflammation patient is found patient_max <- 0 # index (row number) for this patient in this file average_inf_max <- 0 # value of the average inflammation score for this patient for (f in filenames) { dat <- read.csv(file = f, header = FALSE) dat.means <- apply(dat, 1, mean) for (patient_index in 1:length(dat.means)){ patient_average_inf <- dat.means[patient_index] # Add your code here ... } } print(filename_max) print(patient_max) print(average_inf_max)Solution
# Add your code here ... if (patient_average_inf > average_inf_max) { average_inf_max <- patient_average_inf filename_max <- f patient_max <- patient_index }
Comparing inflammation across trials
We want to compare how inflammation for an individual patient changes across trials. Write a script that plots inflammation for the first patient from each trial (i.e. from each file) in the same line graph.
Solution
# Indicate the patient (aka row) number that we want to extract patient.id <- 1 # First, grab our file list using pattern matching inflam.files <- list.files(path = "data", pattern = "^inflammation-.*.csv$", full.names = TRUE) # We can't just plot away, because we don't know the minimum and maximum values. # First, we should cycle through the files and collect the data we need into # a data frame # Initialize matrix for data collection (numeric matrix with a row for each file # and 40 columns for number of days) trial.data <- matrix(nrow = length(inflam.files), ncol = 40) # Start the for loop to cycle through the files for(i.file in 1:length(inflam.files)) { # this time we use a numeric index # grab file name file.c <- inflam.files[i.file] # read in the current file inflam.c <- read.csv(file = file.c, header = FALSE) # assign selected patient to collection file row trial.data[i.file,] <- as.numeric(inflam.c[patient.id,]) } # Initiate the PDF file to store the graphs pdf(file = paste0("results/inflammation-patient",patient.id,"-by-trial.pdf"), height = 5, width = 5) # look up max and min values for complete day to set plot size y_min <- min(trial.data) y_max <- max(trial.data) # first initiat the plot with no data, but define the size and labels plot(NA, # plot minimum first xlab = "Day", ylab = "Inflammation", # axis labels xlim = c(1,40), ylim = c(y_min, y_max)) # define plot limits # select colors with the rainbow() function col.list <- rainbow(length(inflam.files)) # now that we have the plot limits set, use another for loop to cycle through > > # and plot each trials inflammation for(i.plot in 1:dim(trial.data)[1]) { # index down the number of rows lines(trial.data[i.plot,], # plot current trial col = col.list[i.plot]) # choose current color in list } # finalize the PDF file by turning off the graphics device dev.off()As code becomes more complex, the ways to achieve a given solution multiply. This is just one solution of many possibilities.
Gathering data
There is interest in examining the inflammation data in a more complete analysis. To examine effects of treatments across trials, we first need to collect our data into a single file.
Gather all of the data from the various inflammation files into a single data frame with the appropriate labels indicating where each data point originated. Make sure that your script is robust enough to run on any number of inflammation data files.
Solution
# First, grab our file list using pattern matching inflam.files <- list.files(path = "data", pattern = "^inflammation-.*.csv$", full.names = TRUE) # Start the for loop to cycle through the files for(i.file in 1:length(inflam.files)) { # this time we use a numeric index # grab file name file.c <- inflam.files[i.file] # read in the current file inflam.c <- read.csv(file = file.c, header = FALSE) # name the columns names(inflam.c) <- paste0("Inflammation.D",1:40) # add columns for Patient ID and Trial ID inflam.c <- cbind(Patient.ID = 1:60, Trial.ID = i.file, inflam.c) # if this is the first iteration, the combined file is the same as the current file if(i.file == 1) { inflam.combined <- inflam.c # otherwise, add the rows of inflam.c to the growing combined data frame } else { inflam.combined <- rbind(inflam.combined, inflam.c) } } # save file write.csv(inflam.combined, file = "data/combined-inflammation.csv", quote = FALSE, row.names = FALSE)Is there a more efficient way to do this?
Key Points
There are a number of ways to use logical operators, relational operators, and various functions to ask complex and specific questions in the form of
ifstatements.Two commone ways to index the loop variable in
forloops are to use the formfor(variable in collection)to pull the elements (“variable”) from a vector (“collection”) one at at time, or to use the formfor(i in 1:length(collection))orfor(i in seq_along(collection))to instead pull each index (i.e. location) in a vector one at a time.Use functions such as
applyinstead offorloops to conduct repeated operates on values contained within defined subsets of a data structure.Use
list.files(path = "path", pattern = "pattern", full.names = TRUE)to create a list of files whose names match a pattern.